
Testing Primer Sequence Variations Using AS-PCR to Diagnose the V232D-CFTR Mutation
Danielle E. Hohman1, Oren S. Lerner1, Scott M. Nolan1, Grace A. Kuza1, Zakia Zaman1, Douglas B. Luckie1,2*
1Lyman Briggs College, Michigan State University, East Lansing MI, USA
2Department of Physiology, Michigan State University, East Lansing MI, USA
Abstract
There are over 1,700 mutations of the CFTR gene that lead to the autosomal recessive disease cystic fibrosis (CF). Of these, the V232D mutation is a rare condition caused by a single nucleotide polymorphism at the 232nd codon. At the mutation site, a T→A nucleotide change causes GTC (valine), to become GAC (aspartic acid). This promotes the formation of non-native hydrogen bonds, which lead to a misfolding of the CFTR protein. To increase the specificity of detecting the V232D mutation of CFTR, an array of allele specific polymerase chain reaction (AS-PCR) strategies were tested. Preliminary control experiments were conducted by analyzing commercially purified genomes and human genomic DNA isolated from somatic buccal cells using chelex resin. Primers were developed to compare amplification stability versus nucleotide sequence. Applying the Yaku group’s strategy, we hypothesized that a primer designed with the V232D SNP nucleotide mismatch at the second base from the 3’ end would be more effective at diagnosing the V232D mutation of CFTR than a primer with a mismatch at the third base from the 3’ end due to steric strain principle limiting false positive results. Final analysis yielded data to support the function of our mutant seeking primers on mutant DNA. Yet further studies are needed to investigate mismatched nucleotide placement versus annealing and polymerization.
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease that affects the body’s airway secretory exocrine glands. Its symptoms most strongly manifest in the lungs, liver, and pancreas. CF is caused by mutations of the CFTR (cystic fibrosis transmembrane conductance regulator) gene located on chromosome 7. Many of the mutations associated with CF lead to the production of faulty CFTR proteins, which normally serve as chloride channels on the apical surface of epithelial cells (Grove et al., 2009). If a protein's function is impaired, chloride ions cannot be correctly exchanged across the membrane, leading to the salt imbalance and change in composition of mucus. The presence of excess mucus impairs secretory function and, in the airway, can leave the affected organs prone to infection, altering respiratory function (Loo et al., 2012).
The most common CF mutation is ΔF508, accounting for approximately 70% of all CF cases. At the genetic level, ΔF508 causes an error in the folding of the CFTR protein (Caldwell et al., 2011). Another mutation that leads to a misfolding of the CFTR protein is V232D. This mutation is infrequent and often leads to a milder case of cystic fibrosis (refSNP ID: rs397508783). V232D is found on exon 6a of the CFTR gene, altering the 232nd amino acid in the coding sequence. The amino acid valine (GTC) is mutated to an aspartic acid (GAC). It is postulated once this change occurs, the aspartic acid, located on transmembrane region alpha helix 4, creates a non-native hydrogen bond with the 207th amino acid, glutamine, located on transmembrane 3. This hydrogen bond leads to a misfolding of the CFTR protein, which may disrupt CFTR function due to altered protein-lipid interactions (Fernández-Lorenzo et al., 2018; Rath et al., 2009).
Mutant DNA can be amplified for further study with the use of polymerase chain reaction (PCR) (Newton, 1999). For this experiment, the PCR process was used to amplify wild-type CFTR and mutant-type V232D CFTR DNA. Through the use of allele-specific PCR and comparative gel electrophoresis, a distinction can be visualized between the V232D mutant and wild-type specimens to aid in diagnosis as well as additional research (Rath et al., 2009). The central focus of the study was to compare variations in DNA sequence of PCR primers allowing greater accuracy in detection of the mutation.
Hidenobu Yaku found that AS-PCR assays designed to detect single nucleotide polymorphisms (SNPs) could be improved by the use of additional intentional mismatches (Yaku et al., 2008). In this study our goal was to test a similar strategy. We tested the impact of movement in placement of SNP mismatches at the 3'end of primers, to determine if this might reduce the rate of false positives. A primer designed with a nucleotide mismatch on the second annealing site from the 3’ end might be more effective at diagnosing the V232D mutation of CF than a primer with a mismatch on the third annealing site from the 3’ end due to the steric strain principle reducing false positive results. Steric strain refers to a highly unfavorable bonding configuration that is energetically unlikely to occur due to electron repulsion (Yaku et al., 2008). To address the research question, four unique primers were designed to amplify wild as well as mutant-type CFTR alleles. Human genomic DNA was purified from buccal cells while a sample of V232D mutant-type genomic DNA was obtained from Dr. Charles Deber at The Hospital for Sick Children in Toronto. Two of the designed primers, forward-two (F2) and forward-three (F3) were mutant-seeking primers with SNP nucleotide mismatches on the second and third nucleotide from the 3’ end respectively. It was predicted that the F3 primer would bubble around the mismatched base pair and permit amplification. This is because the two remaining 3’ nucleotides would likely still bind to the antisense strand allowing Taq polymerase to initiate replication even though the primer sequence was a mismatch, giving a false positive result. It was hypothesized that amplification of the F2 primer would be more disrupted due to the steric pressure of the mismatched nucleotide being in closer proximity to the 3’ end of the primer and false positive results would be minimized.
Methods
Materials
Human genomic DNA samples were obtained from a solution of basal mucosa taken from male and female human subjects’ epithelial cells with wild-type CFTR gene by the use of Chelex-100 Resin from Sigma Inc. V232D-CFTR mutant-type human genomic DNA was obtained from Dr. Charles Deber from The Hospital for Sick Children in Toronto, Ontario, Canada. DNA sequences of control primers were obtained from Ana E. Fernández-Lorenzo of the Department of Pediatrics at Hospital Teresa Herrera, Coruña, Spain.
Primers
The forward primers were designed to anneal to the antisense strand, whereas the universal reverse primer annealed to the sense strand (Fernández-Lorenzo, 2018). The wild-type seeking primers targeted a DNA segment that amplified a 1046bp long nucleotide fragment beginning on exon 6a of CFTR and extending to intron between exon 6a and 6b, and the mutant seeking primers amplified 1046 (F2) and 1045 (F3) products beginning on exon 6a and extending to intron between exon 6a and 6b. (Choi, 2014). Three primers were created to amplify the target strand of DNA. A universal reverse primer: 5’-GTAAATGCCTCCTATGTGCCAGAC-3’. In addition, three forward primers were designed, one to seek wild-type DNA sequence (5’-TGTGGACTTGGTTTCCTGATAGTC-3’) and two to seek V232D mutant-type DNA with respective manipulated mutation sites on the 3’ end of the primer, either the 2nd (F2) or 3rd (F3) nucleotide change (5’-TGTGGACTTGGTTTCCTGATAGAC-3’ / 5’-GTGGACTTGGTTTCCTGATAGACC-3’). The annealing temperature of our primers was initially predicted using the melting point equation: TM=64.9?+41((G+C-16.4)/(A+T+G+C))? (Wallace, 1979).
Polymerase chain reaction
To conduct PCR, a 50μl cocktail was created using 38.5μlof nuclease free H2O, 5.0μlof 10X HIFI PCR buffer (200 mM Tris-HCl (pH 8.4) and 500 mM KCl, from Invitrogen), 1.5μl of 25mM MgCl2 and 1.0μl of each; 10mM dNTP’s, 100μM forward primer, 100μM reverse primer, 1ug/μl DNA template, and 5U/μl (units of activity per microliter) Taq polymerase. A T100 Bio-Rad thermal cycler was used. Set at a ramp of 4? per second and initial denaturing temperature of 95? for 3 minutes. Cycles began with a denaturing temperature of 95? for 30 seconds. An annealing stage at temperatures dependent on the primer properties followed and lasted for 45 seconds. The reverse primer was found to have a melting temperature of 57.38? and the forward primers to have melting temperatures of 57.38? (F1), 55.67? (F2), 57.38? (F3). To complete the cycle, a polymerization temperature of 72? was selected for one minute to optimize Taq polymerase function and until retrieved from the device, this cycle was set to repeat 30 times after which the samples were set to an indefinite refrigeration stage at 4? (Clark, 2013).
Agarose gel electrophoresis
Once polymerized, each PCR sample was analyzed using gel electrophoresis. A gel with a volume of 60ml was created from 57ml of dH2O, 0.48gof agarose powder, 3ml of 20X Lithium Borate buffer, and 0.5-1ml of SYBR-safe (10,000X concentrate) (Lee, 2012). In each well, 2μl of (6X): Bromophenol blue-based gel loading dye with SDS and EDTA or the respective DNA ladder (1Kb+ or 100bp) was then pipetted into the specified wells of the gel. The gel was run at 300 volts for 15 minutes (Lee, 2012). Gel was analyzed under UV light and photographed using a Bio-Rad Gel Doc EZ Imager. Placement of DNA fragments within the gel were compared to the fragment separation of DNA ladder and analyzed for size on semi-log plot.
E. coli control
An E. coli control was used regularly to assure that reagents continued to perform as expected. This control verified the concentrations and conditions used in PCR and gel electrophoresis and yielded a known result. PCR was conducted with a cocktail of 38.5μl H2O, 5μl 10X PCR HIFI buffer, 1μl 10mM dNTP,1.5μl 25mM MgCl2, 1μl each; forward and reverse primer (8F: 5’-AGAGTTTGATCCTGGCTCAG-3’ TM = 51.78? / 1512R: 5’-ACGGTTACCTTGTTACGACTT-3’ TM= 49.73?), 1μl DNA template, and 1μl Taq polymerase. PCR was run in the T100 Thermocycler, an initial denaturation stage occurred for 3 minutes at 95?. Denaturation occurred for 30 seconds at 95?. Annealing occurred at 47? for 30 seconds. Polymerization occurred for 1 minute and 30 seconds at 72?. Final polymerization occurred at 72? for 5 minutes. This repeated for 25 cycles and generated about 34 million copies of the target strand. E. coli testing served as a positive control for additional PCR diagnostic assay.
Wild-type genomic DNA purification and controls
Purified genomic DNA samples from human wild-type epithelial cells were obtained with the use of Chelex-100 resin and experimentally assessed over multiple trials. The Chelex-100 was diluted to generate a 10% Chelex solution from 0.5g of Chelex-100 resin and5mlof H2O. Wild-type buccal cells from human-cheek swabs were mixed with 10% chelex solution and centrifuged 14k rpm for 2 minutes. In order to lyse the cells and release the DNA, samples were transferred to a 100? sand bath for 5 minutes, mixed, and returned for another 5 minutes. The DNA supernatant was separated from the cell debris and chelex beads by centrifugation for 5 minutes at 8k rpm. The supernatant, containing DNA, was assessed from DNA concentration. Samples of 2µl from each trial were placed in a multi-well adapter and the 260/280nm UV wavelength absorbance was assessed within a Gen 5 Epoch™ microplate spectrophotometer.
Results
Annealing Temperature
When designed the mutant forward primer 3 (F3, Figure 1) and the universal reverse primer were initially calculated to have predicted annealing temperatures of 57.38 ? and the wild-type forward primer 1 (F1) and mutant-type primer 2 (F2) to have annealing temperatures of 55.67 ?. Through experimental testing the binding temperature 53? was found optimal in an effort to ensure the temperature was low enough for successful binding of all primers. This temperature also coincides with the ideal annealing temperature found in previous research (Fernández-Lorenzo, 2009).

Figure 1: Visual representation of PCR on targeted segments of wild-type and mutant-type (V232D) DNA of the human CFTR genome. Four primers were designed to amplify target sequences of wild-type and mutant-type CFTR DNA. A universal reverse primer coded: 5’-GTAAATGCCTCCTATGTGCCAGAC-3’(TM=57.38?)was designed to anneal to the intron between exon-6a and exon-6b on the CFTR gene. The first forward primer coded: 5’-TGTGGACTTGGTTTCCTGATAGTC-3’ (TM=55.67?) was designed to anneal to the middle of exon-6a, on the wild-type sequence, creating a target strand 1046 base pairs in length. Two more forward primers were designed for the mutant-type V232D DNA and were coded, forward primer 2: 5’-TGTGGACTTGGTTTCCTGATAGAC-3’ (TM=55.67?) and forward primer 3: 5’-GTGGACTTGGTTTCCTGATAGACC-3’ (TM=57.38?).
Band Length
Control PCR on E. coli regularly yielded bands of approximately 1,504 bp. The size calculated by semi-log analysis frequently differed from the expected band length by only 2 bases (Figure 2). Frequently there was also some streaking and E. coli genomic DNA observed in the well. Purified genomic wild-type DNA when analyzed through a gel electrophoresis yielded, bright bands in the wells (Figure 3) providing qualitative visual data supportive of the expected length of genomic chromosomal human DNA. Concentration and purity of genomic DNA was also assessed with UV spectrophotometry. Control PCR on purified human DNA samples using published primers was performed to support presence of intact CFTR alleles (Fernández-Lorenzo, 2018). After running control PCR tests with published primer sequences versus genomic DNA, gel electrophoresis identified a DNA band of 385 bp as expected for wild type CFTR template (Figure 4). The band was lighter in comparison to the bright E. coli band on the same gel. A gel was run using six different PCR cocktails, to compare the designed primers versus wild-type and mutant template DNA. When run versus wild-type DNA template only one band appeared compared to the three bands versus mutant V232D-CFTR DNA template (Figure 5). A band of 1083 base pairs for the wild-type DNA was amplified using the F1 primer, very near the expected length of 1046. There appears to be genomic material remaining in the well and the band is light compared to others. The wild-type template versus the F2 primer and the F3 primer did not yield bands. This was supportive that the mutant-seeking primers (F2 & F3) did not bind wild-type CFTR allele nonspecifically. The mutant template DNA, which likely contained both wild-type and V232D-CFTR alleles, was amplified by the F1, F2, and F3 primers; all yielding bands of 877 base pairs. There was evident streaking for all of them, and the mutant DNA band amplified with F1 primer was much lighter than the fairly luminescent mutant DNA bands with F2 and F3 primers. These results were not as supportive as those observed when the forward 2 and 3 primers were applied versus wild-type DNA (Figure 6).
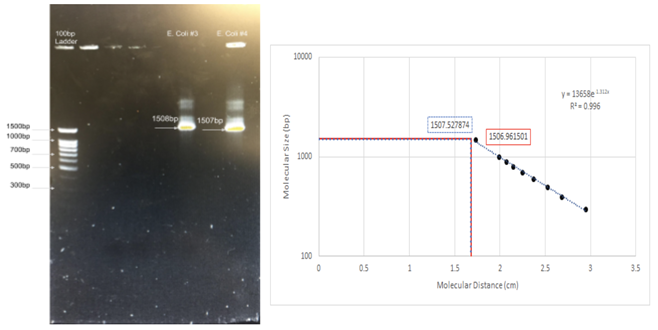
Figure 2: Results of Polymerase Chain Reactions (PCR) on the E. coli genome. Regular application of E. coli testing served as a positive control for the PCR reagents and tools used in developing a diagnostic assay. The forward primer 8F: 5’-AGAGTTTGATCCTGGCTCAG-3’ (Tm: 51.78?); and reverse primer 1512R: 5’-ACGGTTACCTTGTTACGACTT-3’ (TM: 49.73?) were used to amplify part of the E. coli genome. As seen on the semi-log graph, bands were calculated to be 1,506 bp and 1,507 bp for two E. coli controls, nearly exactly the expected length of 1504.
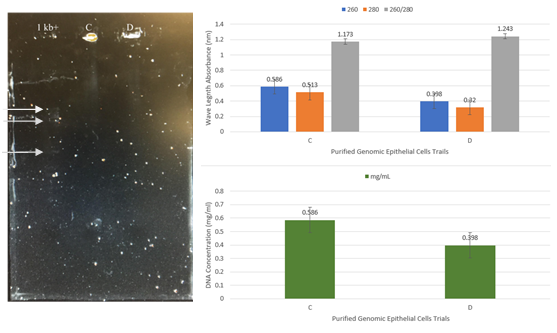
Figure 3: Analysis of purified genomic DNA from human cheek epithelial cells. Two samples of equal volumes were compared using a microplate UV spectrophotometer as well as agarose gel electrophoresis for quantitative and qualitative analysis of human genomic DNA samples. Purified DNA samples from wild-type epithelial cells were obtained with the use of 10% Chelex resin and experimentally assessed over multiple trials (A-E). Trials C and D from spectrophotometry showed the highest value and were evaluated further with gel electrophoresis. As expected, samples remained in wells during electrophoresis due to the large size of genomic DNA.
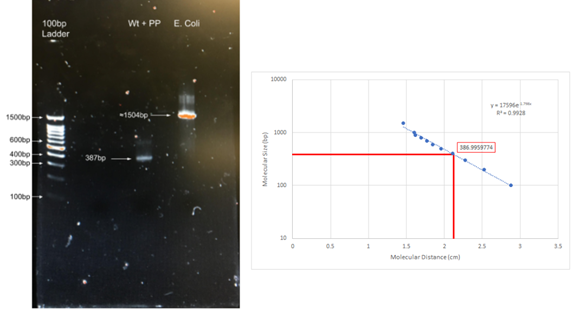
Figure 4: Results of PCR performed on wild-type DNA with control published primers (PP). PCR was performed on purified wild-type DNA, obtained from human saliva buccal cells, using the forward primer, 5’- TTAGTGTGCTCAGAACCACG -3’, and the reverse primer, 5’-CTATGCATAGAGCAGTCCTG -3’. Using the exponential trend line on the graph, it was calculated that the band was 387 base pairs, nearly identical to expected length was 385 bp. A 1504 bp E. coli control band is also present. This test served as an additional regularly applied positive control for the presence of the CFTR gene in purified human genomic DNA samples.
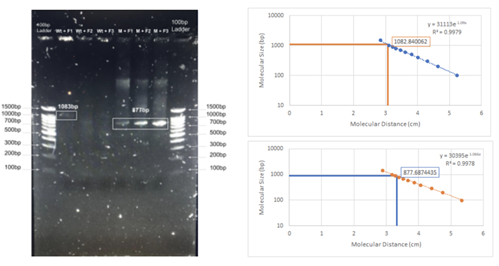
Figure 5: Results of PCR with designed primers on wild-type DNA and mutant DNA. For each cocktail, the universal reverse primer was used 5’-GTAAATGCCTCCTATGTGCCAGAC-3’. For WT+F1 and M+F1 the forward primer 5’-TGTGGACTTGGTTTCCTGATAGTC-3’ was used. For WT+F2 and M+F2 forward primer 5’-TGTGGACTTGGTTTCCTGATAGAC-3’ was used. For WT+F3 and M+F3 the forward primer 5’-GTGGACTTGGTTTCCTGATAGACC-3’ was used. Both standards used were 100bp ladders. The WT+F1 produced a light band calculated to be 1083 base pairs (expected product 1046 bps). Additionally, some genomic DNA appears to remain in the WT+F1 well. As expected the WT+F2 and WT+F3 did not yield detectable amplification. The M+F1, M+F2 and M+F3 all resulted in bands with calculated lengths of 877 base pairs. M+F1 banding is evidence of a heterozygous DNA sample, M+F2 was expected to have 1045 bp and M+F3was expected to have 1046 bp.
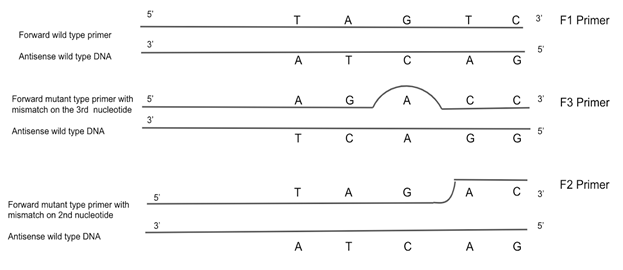
Figure 6: Last five nucleotides of forward type primers compared to the wild-type antisense strand. Three different primers were tested in developing a method of diagnosis for the V232D cystic fibrosis mutation. The universal reverse primer remained constant: 5’-GTAAATGCCTCCTATGTGCCAGAC-3’. The F1 primer was a forward wild-type-seeking primer: 5’-TGTGGACTTGGTTTCCTGATAGTC-3’. This primer contained no mismatch, therefore it was designed to anneal to the wild-type DNA. The F3 primer was designed to test for sequence preference: 5’-GTGGACTTGGTTTCCTGATAGACC-3’. The F3 primer contained nucleotide mismatch on the 3rd nucleotide from the 3’ end of the primer. The F2 primer was designed to anneal to the mutant-type DNA: 5’-TGTGGACTTGGTTTCCTGATAGAC-3’. The mutant nucleotide mismatch is 2 nucleotides from the 3’ end, which might allow it to bind to mutant-type DNA but not bind to the wild-type DNA because there is not enough room for Taq to bind the 3’ end with the mismatch closer to the end. The F2 primer was predicted to work the best to diagnose the V232D cystic fibrosis mutation.
Discussion
Cystic fibrosis (CF) is the most common autosomal recessive disease in Caucasians (Ernst, 2011). This lethal genetic disease presents itself with multisystemic failure mainly affecting the respiratory system, gastrointestinal tract and reproductive organs (Welsh, 2001). The genetic base for the disease is due to the alteration of the cystic fibrosis transmembrane conductance regulator (CFTR) gene which primarily encodes for a chloride channel protein essential for ion transport (Rommens, 1989). However, there are large numbers of CF-related mutations where the disease is caused by a misfolding in the chloride channel (Caldwell, 2011). With CF, alteration or loss of function of the protein leads to an imbalance of ion exchange, resulting in dehydration of the mucous secretion in many organs, most notably within the respiratory tract (Welsh, 2001). V232D is a mutation of the CFTR gene that exhibits defective folding and trafficking (Caldwell, 2011). The CFTR gene consists of 27 exons, spanning more than 180,000 base pairs, located on exon 6a is the V232D mutation. This genetic mutation is a change from a Valine (GTC) to Aspartic Acid (GAC) within exon 6a, nucleotide 827, in the protein sequence at the 232nd Amino Acid. The folding defect caused by the V232D mutation appears to be due to the introduction of a charged residue into a region of CFTR that is embedded in the lipid bilayer of the endoplasmic reticulum membrane (Rath, 2009). The V232D mutation is considered to lead to somewhat fewer extreme symptoms; however mild symptoms can nonetheless cause significant damage to livelihood and life-expectancy (Fernández-Lorenzo, 2018). It was hypothesized that a primer designed with a nucleotide mismatch on the second annealing site from the 3’ end (55,399 bp to 55,423 bp) would be more effective at diagnosing the V232D mutation than a primer with a mismatch on the third annealing site from the 3’ end because of steric strain limiting false positive results.
Original Predictions
In addition to control primers, four different primers were designed to conduct the experiment. These designed primers consisted of a universal reverse primer, which was the same for both wild-type and mutant samples, as well as a wild-type seeking forward primer and two mutant-seeking forward primers (Kwok, 1994). One mutant-seeking forward primer (F2) was designed so the single nucleotide mutation, the SNP change in the nucleotide sequence, at annealing would be located two nucleotides from the 3’ end of the primer, whereas the other mutant-seeking forward primer placed the SNP mismatch on the third nucleotide from the 3’ end (F3). The forward wild-type primer (F1) was used to distinguish between the CFTR gene respective mutant and wild-type DNA samples in accordance with the single nucleotide mismatch. Further, it was predicted that when F1 was used in PCR versus mutant DNA template, given the DNA template is from a patient heterozygous for V232D-CFTR allele, the wild-type-seeking forward primer would not anneal to one chromosome but would to the other, and thus PCR would produce some product, and F2 or F3 primers versus mutant DNA template would also yield a band of approximately 1046 bp (Sommer, 1989). Additionally, it was predicted that when the wild-type genome was present as DNA template PCR would again result in a band of 1046 bp when using the F1 wild-type seeking primer, no band using F2, but perhaps a nonspecific amplification using F3. The mismatch in F3 is shifted in far enough from the 3' end of the primer that Taq polymerase might more likely successfully bind to the 3’ end and amplify due to lower steric strain, whereas when using F2, it was hypothesized that Taq polymerase would not be able to bind at the 3’ end and the PCR would not amplify a product (Sommer, 1989).
Ultimate Findings
The optimal universal annealing temperature for the designed primers used was found to be 53?, which is 2? less than the lowest melting temperature of 55?. This allowed proper denaturing and annealing without melting the primers themselves.
Amplification of E. coli bacteria occurred using 8F and 1512R primers in a PCR cocktail that supplied with MgCl2. A band of 1504 bp was expected and seen in the semi-log graph in Figure 2 (McCauley, 2018). The E. coli test regularly acted as a positive control as it produced a band of the expected base pair length which confirmed the PCR cocktail components and concentrations.
The successful purification of genomic DNA was assessed in two ways: visualized in gel electrophoresis, and absorbance in UV spectrophotometry to detect DNA and RNA content. Amplification of human genomic DNA from buccal cells using published primers also served as a positive control for wild-type results. A band of 385 bp was expected, and was present in visualized gel and calculated size in semi-log plot presented in Figure 4. The wild-type genomic band was faint but present. This band served as a methodological control as there was adequate purified genomic DNA from the samples for further experimentation.
Amplification of wild-type genomic DNA with the three different forward primer sequences was successful. When amplified with F1 primers a faint band found at 1046 bp was present. Neither the F2 or F3 primers, both of which were complementary to the mutant DNA, annealed to the wild-type DNA which is why lanes three and four lack bands (Hellman, 2007). The original concern, that F3 primer sequence could bind to our wild-type DNA giving a false positive, did not occur. As designed, all three forward primers yielded a band of 877 bp when amplified with the human genomic DNA containing the V232D-CFTR allele. Both F2 and F3 primers were expected to yield bands because they were complements of mutant DNA. Even though the received band was 877 bp, it was expected to be 1045 for F2 and 1046 for F3. They were shorter than expected due to either non-specific amplification or potential DNA degradation.
Future Studies
Traditional PCR and agarose gel electrophoresis were used in this study to evaluate primer sequence impact and provided little evidence that the F2 primer is more discriminatory than F3 primer as hypothesized. In future studies, more modern techniques such as real-time PCR, or MassARRAY analysis as well as homogeneous methods using intercalating dye & High Resolution Melting (HRM) and real-time PCR (e.g. with TaqMan probes) could permit much greater number of samples to be rapidly tested to provide more detail detecting homozygous versus heterozygous genotypes. In addition, it would be useful to sequence the 877bp products generated since the WT+F1 experiment generated the predicted band size but M+F1 did not as well as useful to expand the testing to evaluate control DNA templates from patients heterozygous versus homozygous for V232D-CFTR.
Although V232D may be specific to particular geographic areas and population groups, Caucasian or of Spanish origin, it should not be understood as a mild mutation. The symptoms of mutation V232D still comprise of impaired respiratory function and colonization by resistant microorganisms, and such conditions cannot be considered typical of a mild mutation (Fernández-Lorenzo, 2018). Studies with newborn screening, using the detection of the V232D mutation, enables CF to be diagnosed before the onset of symptoms and lung damage.
References
- Caldwell RA, Grove DE, Houck SA, et al. Increased folding and channel activity of a rare cystic fibrosis mutant with CFTR modulators. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2011; 301(3): L346-L352.
- Choi MY, Cardarelli L, Therien AG, et al. Non-Native Interhelical Hydrogen Bonds in the Cystic Fibrosis Transmembrane Conductance Regulator Domain Modulated by Polar Mutations. Biochemistry. 2004; 43: 8077-8083.
- Clark DP, Pazdernik NJ. Molecular Biology - 2nd ed. Chapter 6 “Polymerase Chain Reaction”. 2013.
- Ernst MM, Johnson MC, Stark LJ. Developmental and psychosocial issues in Cystic Fibrosis. Child and Adolescent Psychiatric Clinics of North America. 2011; 19: 2.
- Fernández-Lorenzo AE, Moreno-Álvarez A, Colon-Mejeras C, et al. V232D mutation in patients with cystic fibrosis: Not so rare, not so mild. Li. Y, ed. Medicine. 2018; 97(28): e11397.
- Grove DE, Rosser MF, Ren HY, et al. Mechanisms for Rescue of Correctable Folding Defects in CFTRF508. The American Society for Cell Biology. 2009; Vol. 20: 4059–4069.
- Hellman LM, Fried MG. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nature protocols. 2007; 2(8): 1849-61.
- Hervieu G. A quick and safe method for destaining Coomassie-Blue-stained protein gels. Technical Tips Online. 1997; Vol. 2: 94-96.
- Kwok S, Chang SY, Sninsky JJ, et al. A Guide to the design and use of mismatched and degenerate primers. Genome Research. 1994; 3: S39-S47.
- Lee PY, Costumbrado J, Hsu CY, et al. Agarose Gel Electrophoresis for the Separation of DNA Fragments. Journal of Visualized Experiments: JoVE. 2012; (62): 3923.
- Loo TW, Bartlett MC, Shi L, et al. Corrector-mediated rescue of misprocessed CFTR mutants can be reduced by the P-glycoprotein drug pump. Biochemical Pharmacology. 2012; Vol. 83 Iss. 3: 345-354.
- McCauley B. DNA Electrophoresis Method. Bio 6B Cells and Molecular Biology. 2018.
- Newton CR, Graham A. PCR, second edition. Hong Kong Medical Journal. 1999; Vol. 5 No. 3: 192.
- Rath A, Glibowicka M, Nadeau VG, et al. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. National Academy of Sciences. 2009; Vol 106 no. 6: 1760-1765.
- Rommens JM, Iannuzzi MC, Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989; 245: 1059-65.
- Sommer R, Tautz D. Minimal homology requirements for PCR primers. Nucleic Acids Research. 1989; 17(16): 6749.
- Wallace RB, Shaffer J, Murphy RF, et al. Hybridization of synthetic oligodeoxyribonucleotides to phi chi 174 DNA: the effect of single base pair mismatch. Nucleic Acids Research. 1979; 6(11): 3543-3557.
- Welsh MJ, Ramsey BW, Accurso F, et al. editors. Cystic fibrosis: metabolic and molecular bases of inherited disease. Cystic Fibrosis 8th ed. New York: McGraw-Hill. 2013; 5121–88.
- Yaku H, Yukimasa T, Nakano S, et al. Design of allele-specific primers and detection of the human ABO genotyping to avoid the pseudopositive problem. US National Library of Medicine NAtional Institution of Health. 2008; 29(20): 4130-40.