
PEX6: An Imaging Overlap Between Peroxisomal and Lysosomal Storage Diseases
César Augusto Pinheiro Ferreira Alves1,3*, Luisa Norbert Simonsen2, Jonathan Rodrigues1, Isabella Peixoto de Barcelos2, Clarissa Bueno2, Ramon Moura Dos Santos1, Fernando Kok2,4, Leandro Tavares Lucato1
1Neuroradiology Section, Hospital das Clínicas da Universidade de São Paulo, Brazil
2Neurogenetics Unit Neurology Department, Hospital das Clínicas da Universidade de São Paulo, Brazil
3Division of Neuroradiology, Department of Radiology, Children’s Hospital of Philadelphia, Philadelphia, Pennsylvania, USA
4Mendelics Genomic Analysis, São Paulo, SP, Brazil
Abstract
Peroxisomal disorders are a group of expanding genetic diseases divided into two major categories: peroxisome biogenesis defects (Zellweger spectrum disorder), and single enzymatic defects. Disorders of Peroxisome Biogenesis occur when there are biallelic pathogenic variants in any of the 13 PEX genes, which code for the peroxins, proteins required for peroxisome biogenesis. This group of disorders includes two distinct phenotypes: Rhizomelic Chondrodysplasia Punctata Type-1 and Zellweger Spectrum Disorders (ZSD), of which Zellweger syndrome is the most severe, neonatal adrenoleukodystrophy is intermediate, and infantile Refsum is the mildest. The spectrum’s most frequent defects are observed in the proteins PEX1 and PEX6, and the most common clinical presentation is Zellweger spectrum, which is often associated with craniofacial dysmorphism with neurologic abnormalities. Typically, the neuroimaging pattern shows several malformative features, including a range of cortical gyral abnormalities such as microgyria and pachygyria, and impairment of the myelination. Nevertheless, we report two siblings with peroxisomal disorder, with unexpected leukodystrophy pattern of the brain mimicking lysosomal storage disease, with classical imaging features of Krabbe disease on brain magnetic resonance image.
By whole exome sequencing, we identified two pathogenic variants in compound heterozygosity in PEX6: Chr6:42.933.455 C>T (c.2435G>A), and Chr6:42.935.188 C>T (c.1802G>A). Thus, a final diagnosis of peroxisome disorder was confirmed. The index cases highlight the importance of considering peroxisome disorders as a differential diagnosis for patients with imaging features that resemble Krabbe disease.
Introduction
Peroxisome biogenesis disorders (PBD) are rare autosomal recessive diseases with early-onset, severe systemic involvement, poor outcome, and premature mortality1,2. It involves two groups of diseases: 1- Zellweger Spectrum Disorders (a clinical continuum from Zellweger syndrome - the most severe, Neonatal Adrenoleukodystrophy - the intermediate, and Infantile Refsum - the mildest presentation), and 2- Rhizomelic Chondrodysplasia Punctata Type-11,2.
The phenotypic description could be wider, with 1- a very mild form of PBD with isolated progressive ataxia, 2- Heimler’s syndrome (sensorineural hearing loss, imperfect amelogenesis, nail abnormalities, and retinal pigmentation); and even a more 3- severe neonatal onset with dysmorphic features, encephalopathy, and liver dysfunction3,4.
Zellweger spectrum disorder (ZSD), also referred to as a cerebrohepatorenal syndrome, corresponds to the most frequent and most severe phenotype of the presentation, commonly associated with several malformations, particularly affecting the central nervous system1,2. However, considerable variability of disease severity related to the biochemical and molecular basis of peroxisome disorders can occur, many of them with a scarce analysis in the literature and without genetic correlation2. This disease is secondary to genetic defects in peroxins (Pex), proteins associated with peroxisome biogenesis. The most common genetic defects are observed in PEX1 (60.5% of the cases) and PEX6 (14.5%)5.
Hereby, we report two siblings with a progressive motor and cognitive decline, going through several years of an extensive clinical, laboratory, and imaging studies follow-up. Interestingly, brain magnetic resonance imaging (MRI) studies have demonstrated extensive involvement of the white matter, highlighting several imaging features mimicking the leukodystrophy pattern of the description of Krabbe disease6,7.
Both children had normal galactocerebrosidase levels, ruling out the diagnosis of Krabbe and had also ruled out saposin A deficiency diagnosis. After a whole exome sequencing (WES), two heterozygous variants were found in the PEX6: p.(Arg812Gln) and p.(Arg601Gln). Therefore, a diagnosis of peroxisome disorder was established.
Case 1
A full-term 9-year-old female, the first child of non-consanguineous parents, was born from a spontaneous vaginal delivery, after an uneventful pregnancy. Early neurologic development was normal until 2 years and 6 months, when she started focal farmacoresponsive epilepsy, progressive cognitive delay, incoordination, and gait ataxia. Brain MRI performed at the age of 4 years and 6 months showed symmetric abnormalities in the periventricular and deep white matter, with sparing of subcortical U fibers. The white matter lesions compromised also the splenium of the corpus callosum, the posterior limbs of the internal capsules (corticospinal tracts), cerebral peduncles, pons, pyramids, cerebellar hemispheres, and dentate nuclei (figure 1A-D). Moreover, areas of normal signal intensity of the white matter resembling a “tigroid” pattern on axial images were noted, characterized by T2-hypointense dots and stripes in the deep white matter (figure 1E). Additional imaging features included diffuse thalamic involvement, enlargement and contrast enhancement involving the intracranial portion of both optic nerves and chiasm; and also, enhancement of both oculomotor nerves in the interpeduncular cistern (figure 1F-G). These neuroimaging features, in accordance with clinical findings, raised the suspicion of galactocerebrosidase deficiency (Krabbe disease). However, this diagnosis was not confirmed by laboratory and genetic tests.
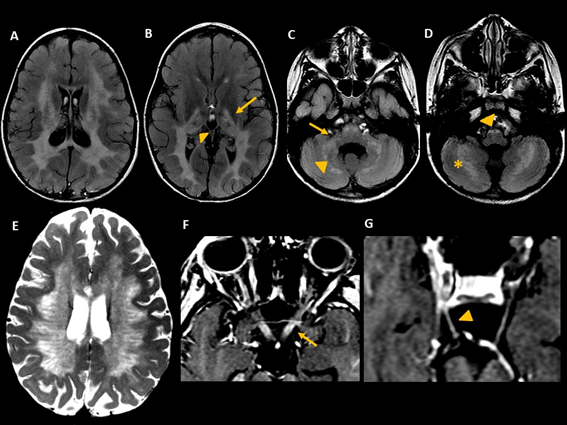
Figure 1: MRI from case #1. Axial FLAIR images (A, B, C, D) demonstrate hyperintensity in periventricular and deep white matter, extending to the posterior limbs of the internal capsules (arrow, B), thalamic involvement (arrowhead, B), middle cerebellar peduncles (arrow, C), pyramids (arrowhead, D), cerebellar hemispheres (asterisk, D) and dentate nuclei (arrowhead, C). Axial T2-weighted image (E) shows tigroid appearance of the white matter. Postcontrast axial T1-weighted images (F and G) disclose enlargement and enhancement of both optic nerves (arrow in F) and enhancement of both oculomotor nerves (arrowhead in G).
After a more comprehensive genetic analysis with WES, two heterozygous variants were found in PEX6: variant 1 - Chr6:42,933,455C>T (GRCh37 / Hg19), c.2435G>A (ENST00000304611), p.(Arg812Gln); variant 2 - Chr6:42,935,188C>T, c.1802G>A, p.(Arg601Gln). Both variants have been reported before in PBD.
Case 2
A 5-year-old male, brother of the former patient (case #1), presented distinct clinical picture, with global developmental delay without regression, facial dysmorphism (epicanthal folds, long philtrum broad nasal bridge, high palate, low set, simplified ears, and hypertrichosis), hypotonia, ataxia and hepatomegaly. Echocardiogram was normal, and ENMG (electroneuromyography) identified axonal motor polyneuropathy. At this time, he performed a brain MRI, which revealed symmetric involvement of the deep WM of the cerebellar hemispheres, notably surrounding the dentate nuclei (figure 2A). There was also symmetric signal abnormality of the bilateral peritrigonal regions, (figure 2B) and a chronic area of a previous hemorrhage in the white matter of left centrum semiovale (figure 2C-D). He was also subsequently diagnosed by Sanger sequencing with the same PEX6 pathogenic variants.
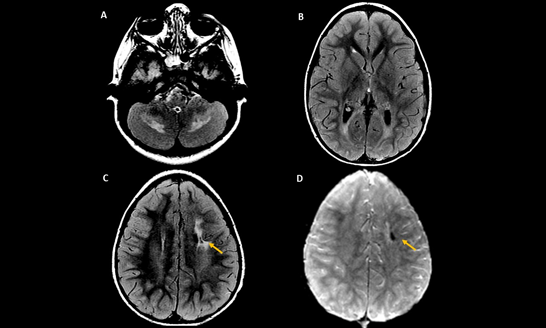
Figure 2: MRI from case #2. Axial FLAIR images (A and B) demonstrate hyperintensity of the deep white matter of the cerebellar hemispheres (A) and bilateral peritrigonal areas (B). Axial FLAIR (C) and susceptibility image sequence (D) show a small chronic hemorrhagic area in the left centrum semiovale (arrows in C and D).
Discussion
In the presenting cases of PBD, the neuroimaging findings suggest lysosomal storage diseases, especially Krabbe disease, and less likely metachromatic leukodystrophy. Typical Krabbe disease MRI features (including symmetrical leukodystrophy with a “tigroid” pattern, selective involvement of the corticospinal tract, enhancement and enlargement of the optic nerves, and enhancement of other cranial nerves) raised this diagnosis as the first hypothesis6,7, despite the clinical correlation of the two-and-a-half-year milestone regression framework more favorable to the latest. Moreover, presence of facial dysmorphism, facial hypertrichosis, global hypotonia and hepatomegaly observed in the younger patient (case 2), did not contribute for the differential diagnosis.
After an extensive follow-up with clinical, laboratory, and imaging studies, by WES we identified two heterozygous variants in the PEX6: p.(Arg812Gln), previously reported as pathogenic in ZSD individuals5, and p.(Arg601Gln), a variant with an allele with the relatively high allele frequency of 0.003 in GnomAD Browser and present in homozygous state in 7 among 138,856 individuals in the same databank . These findings suggested that 2 copies of this variant do not lead to the disease development. This type of variant is called hypomorphic and poses a risk for ZSD when in trans with another pathogenic allele.
The phenotypic variation seen in ZSD is thought to be related to the severity of the underlying PEX gene variants. However both alleles have been associated with mild forms of PBD, being the p.(Arg601Gln) variant previously reported in mild forms of ZSD and the hypomorphic p.(Arg812Gln) reported in seven patients with Heimler syndrome3,4, in comparison with our patient with severe neurodevelopmental regression and leukodystrophy phenotype and no clinical suggestion of PBD. We hypothesize that the prevalence of mild phenotypes secondary to compound heterozygous with the hypomorphic allele is estimated to be higher in patients with clinical symptoms who do not initially hypothesize peroxisomal disease, as in our patients. We also performed a subsequent analyzes in PEX1, searching for another associated pathogenic variant that could justify the severity of the phenotype observed. A more severe phenotype presentation has been previously described in patients that have AP4M1 association. No other pathogenic variants were observed in our patients8.
Regarding imaging analysis, there is also a large variety of patterns associated with PBD, many of them with a scarce analysis in the literature and without genetic correlation5, being the most characterized the most severe end of the spectrum which includes neuronal migration defects, pachygyria, caudothalamic germinolytic cysts, bilateral perisylvian polymicrogyria, and also severe cortical and subcortical atrophy with ventricular dilatation2. Presence of delayed myelination or, less frequently, demyelinating lesions distributed in the periventricular and deep WM with a parieto-occipital gradient, affecting the corpus callosum, posterior limbs of the internal capsules, cerebral peduncles, pons, and pyramids may be also observed2,9.
Krabbe disease, also known as globoid cell leukodystrophy, is a rare autosomal recessive hereditary disease caused by a deficiency of lysosomal galactocerebrosidase (GALC) resulting in demyelination of both central and peripheral nervous system where the commonest presentation is the rapid neurological deterioration typical of the early onset subtype10,11.
Once many diseases can mimic Krabbe disease, the list of differentials diagnosis is extensive such as metachromatic leukodystrophy, X- linked adrenoleukodystrophy, neuronal ceroid lipofuscinosis, GM2 gangliosidoses and other leukodystrophies like vanishing white matter disease6,7,12. Moreover, Krabbe disease with late onset can also mimic other diseases such as multiple sclerosis, and acute disseminated encephalomyelitis11,13.
In this article, we have described two patients with atypical neuroimaging and clinical features due to biallelic pathogenic variants in PEX6.
To date no descriptions of PEX6 pathogenic variants mimicking Krabbe disease have been found in the literature, and we understand that the recognition of this imaging pattern linked with PBD, particularly PEX6, can be helpful in the clinical practice guiding physicians to consider this differential diagnosis in this set of clinical and imaging data. As the prognosis between them is remarkably variable, an accurate diagnosis is essential due to facilitate appropriate treatment and familiar counseling.
This overlap of findings between peroxisomal disorders and lysosomal storage diseases has biochemical substrate in the literature, due the transport of cholesterol between the membranes of peroxisome and endoplasmic reticulum (ER)14,15,16. Also, the accumulation of cholesterol inside the lysosomes of peroxisomal disorders has already been reported in X-linked adrenoleukodystrophy (ABCD1), and Zellweger spectrum disorder (PEX1 or PEX26)17,18,19. Also, the clinical phenotype of the described peroxisomal diseases overlaps with lysosomal storage diseases, such as Niemann-Pick type C 20.
Conclusion
Expand the differential diagnosis of imaging patterns classically defined for Krabbe disease in the context of other genetic and biochemical disorders, including PBD, may help to guide neuroradiologists and pediatric neurologists to the accurate diagnosis in such challenging cases.
These two cases reemphasize that lysosomal disorders, such as Krabbe disease, may be mimicked by other leukodystrophies, and that the phenotypic spectrum of this disease must include peroxisomal disorders in the differential diagnosis.
Acknowledgements
We thank the patients and their families for their involvement.
Conflict of interests
Fernando Kok is medical director and shareholder of Mendelics Genomic Analysis.
References
- Braverman NE, Raymond GV, Rizzo WB, et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 2016; 117(3): 313–321.
- Pfeifer CM, Martinot CA. Zellweger syndrome: Depiction of MRI findings in early infancy at 3.0 Tesla. Neuroradiol. J. 2017; 30(5): 442–444.
- Smith CEL, Poulter JA, Levin AV, et al. Spectrum of PEX1 and PEX6 variants in Heimler syndrome. Eur. J. Hum. Genet. 2016; 24(11): 1565–1571.
- Ratbi I, Falkenberg KD, Sommen M, et al. Heimler Syndrome Is Caused by Hypomorphic Mutations in the Peroxisome-Biogenesis Genes PEX1 and PEX6 [Internet]. The American Journal of Human Genetics 2015; 97(4): 535–545.Available from: http://dx.doi.org/10.1016/j.ajhg.2015.08.011
- Ebberink MS, Mooijer PAW, Gootjes J, et al. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Hum Mutat. 2011; 32(1): 59–69.
- Eluvathingal Muttikkal TJ, Montealegre DR, Matsumoto JA. Enhancement of multiple cranial and spinal nerves in vanishing white matter: expanding the differential diagnosis. Pediatr Radiol. 2018; 48(3): 437–442.
- Muthusamy K, Sudhakar SV, Thomas M, et al. Revisiting magnetic resonance imaging pattern of Krabbe disease - Lessons from an Indian cohort. J Clin Imaging Sci. 2019; 9: 25.
- Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011; 478(7367): 57–63.
- Tran C, Hewson S, Steinberg SJ, et al. Late-onset Zellweger spectrum disorder caused by PEX6 mutations mimicking X-linked adrenoleukodystrophy. Pediatr Neurol. 2014; 51(2): 262–265.
- Mamada N, Nakamagoe K, Shioya A, et al. Adult-onset Krabbe disease presenting as acute hemiparesis and progressive demyelination detected by diffusion-weighted imaging. J Neurol Sci. 2016; 367: 326–328.
- Arenson NE, Heydemann PT. Late-Onset Krabbe’s Disease Mimicking Acute Disseminated Encephalomyelitis [Internet]. Pediatric Neurology. 2005; 33(3): 208–210.Available from: http://dx.doi.org/10.1016/j.pediatrneurol.2005.03.010
- Kohlschütter A. Lysosomal leukodystrophies: Krabbe disease and metachromatic leukodystrophy. Handb Clin Neurol. 2013; 113: 1611–1618.
- Tomás J, Durães J, Lacerda L, et al. Adolescent-onset Krabbe disease with an initial diagnosis of multiple sclerosis and a novel mutation [Internet]. BMJ Case Rep. 2015; 2015Available from: http://dx.doi.org/10.1136/bcr-2015-210625
- Xiao J, Luo J, Hu A, et al. Cholesterol transport through the peroxisome-ER membrane contacts tethered by PI(4,5)P and extended synaptotagmins. Sci China Life Sci. 2019; 62(9): 1117–1135.
- Chu BB, Liao YC, Qi W, et al. Cholesterol transport through lysosome-peroxisome membrane contacts. Cell. 2015; 161(2): 291–306.
- Luo J, Jiang L, Yang H, et al. Routes and mechanisms of post-endosomal cholesterol trafficking: A story that never ends. Traffic. 2017; 18(4): 209–217.
- Berger J, Gärtner J. X-linked adrenoleukodystrophy: clinical, biochemical and pathogenetic aspects. Biochim Biophys Acta. 2006; 1763(12): 1721–1732.
- Jo DS, Park NY, Cho DH. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp Mol Med. 2020; 52(9): 1486–1495.
- Aubourg P, Wanders R. Peroxisomal disorders. Handb Clin Neurol. 2013; 113: 1593–1609.
- Geberhiwot T, Moro A, Dardis A, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis. 2018; 13(1): 50.