
Hemoglobin AE, AO-Arab and SO-Arab Genotypes in Burkina Faso: Hematological Parameters, Genotypic and Allelic Frequencies of Hemoglobinopathies
Marie Simone Traore1,2, Theodora M. Zohoncon1,2,3,4, Paul Ouedraogo3, Abdoul Karim Ouattara1,2, Dorcas Obiri-Yeboah5, Issoufou Tao1,2, Geoffroy Ganane1,2,3, Marius Belemgnegre3, Theodore Boro3, Fabienne Sanou3, Jacques Simpore1,2,3,4*
1Laboratory of Molecular Biology and Genetics (LABIOGENE), University Joseph KI-ZERBO, Ouagadougou, Burkina Faso
2Pietro Annigoni Biomolecular Research Center (CERBA), Ouagadougou, Burkina Faso
3Saint Camille Hospital of Ouagadougou (HOSCO), Ouagadougou, Burkina Faso
4Faculty of Medicine, University Saint Thomas d'Aquin (USTA), Ouagadougou, Burkina Faso
5Department of Microbiology and Immunology, School of Medical Sciences, University of Cape Coast, PMB, Cape Coast, Ghana
Abstract
Introduction: The prevalence of hemoglobinopathies (HbC and HbS) is relatively high in West Africa, especially in Burkina Faso. The objectives of this study were to characterize the hematological parameters and to determine the genotypic and allelic frequencies of patients affected with hemoglobinopathies.
Methods: Hemoglobin electrophoresis was carried out in a total of 7,789 patients attending Saint Camille Hospital of Ouagadougou during the period of study. Among them, hemogram was performed for 1014 patients.
Results: The age of the study population ranged from 1 to 40 years, with a mean of 24.86 ± 12.69 years. The age group 16 to 35 were the most seen at medical center hospital and represented 3,035 out of 7,789 (38.96 %) having performed hemoglobin electrophoresis. The overall hemoglobin electrophoretic profiles revealed 223 SS, 718 SC, 2 SO-Arab, 2 AO-Arab, 3 AE, 152 CC, 799 AS, 1315 AC, and 4575 AA. The relative genotypic frequency was 58.73 % AA, 2.86 % SS and 9.22 % SC while the allelic frequencies were 0.7234 for HbA, 0.1500 for HbC, 0.1261 for HbS, 0.0002 for HbE and 0.0003 for HbO-Arab. The hemogram profiles of patients with major sickle cell syndrome (MSCS) revealed respectively for mean hemoglobin levels, number of red blood cells and mean corpuscular volume a values of 8.19 ± 1.39 g/dL ; 2.97 ± 0.73 1012/L ; 81.86 ± 12.82 fL in SS patients and 10.93 ± 1.68 g/dL ; 4.38 ± 0.77 1012/L ; 70.81 ± 7.11 fL in SC individuals.
Conclusion: The results of the present study are in line with the previous one describing the high prevalence of HbC and HbS hemoglobinopathies in Burkina Faso. Indeed, the genotypic and allelic frequencies of patients with MSCS are increasing due to the accessibility of medical care for sickle cell patients. The study diagnoses for the first-time hemoglobin AE, AO-Arab and SO-Arab genotypes in Burkina Faso.
Introduction
Hemoglobinopathies are the most common genetic diseases in the world. They are divided into two main types, qualitative abnormalities or hemoglobinopathies and quantitative anomalies named thalassemia1. Sickle cell disease is the most common hemoglobinopathy in the world2. It is characterized by the presence of the abnormal hemoglobin S (HbS) in red blood cells, with mutation in β-globin chains3. The gene that codes for hemoglobin is located on the short arm of chromosome 11 at 11p15.5. The βs form is a monogenic disorder caused by single base transition in the sixth codon of the β-globin gene first exon (GAG to GTG) resulting in the substitution of glutamic acid with valine. βc form is induced by G to A transversion (GAG into AAG) leading to a substitution of a glutamic acid with lysine4. There are over 600 known hemoglobin variants5 and the most common in addition to HbS, are HbC, HbE, HbO-arab and HbD-Punjab. People with hemoglobinopathy traits represent 5 % of the world population6,7. Most people affected with the disease live in Africa with a prevalence between 10 and 40%8. Major Sickle Cell Syndromes (MSCS) are due to S homozygosity or heterozygosity associated with a mutation affecting the second chain β namely composite heterozygote such as SC, SD-Punjab, SO-arab, Sβ0 thalassemia, Sβ+ thalassemia9. Some forms of MSCS such as SE, SD-Punjab, S Antilles and SO-arab are rarer2. Individuals with MSCS are often victims of vaso-occlusive crises and are more susceptible to parasitic (malaria) or bacterial (pulmonary, bone and digestive infections) infections10,11. Sickle cell disease related death is estimated at 100,000 black children worldwide each year12. It is first chronic hemolytic anemia in sub-Saharan Africa and a global public health disorder13. Over 70% of children with sickle cell disease worldwide were born in sub-Saharan Africa with a neonatal prevalence of the disease estimated at 2% or more7,10. In Burkina Faso, the prevalence of MSCS is about 8.42%10,14. Sickle cell anemia, responsible for a high rate of consultations, early mortality and school absenteeism, is a major public health problem in Burkina Faso16. Therefore, the country is committed to sickle cell anemia prevention and control with awareness and better monitoring of patients. This, therefore, raises the necessity to update data regularly. The objectives of our study were to characterize the hematological parameters and to determine the genotypic and allelic frequencies of the βS, βC, βE, βO-arab globin genes of patients affected by hemoglobinopathies.
Methods
Samples and patients
The present retrospective, descriptive and analytical study was carried out at Saint Camille Hospital of Ouagadougou (HOSCO), Burkina Faso from April 10, 2017 to August 4, 2019. HOSCO is a benchmark hospital with several services including the department of sickle cell disease monitoring and management. Likewise, that the study of Simpore et al. (2002), the present study population was seen for medical consultations, either systematic surveillance or consecutive to a pathological condition. The study population consisted of people who performed hemoglobin electrophoresis at HOSCO associated or not with hemogram during the period of study regardless of origin, gender, or age. The socio-demographic data (age and gender), and biological data (hemogram, and hemoglobin electrophoresis) were retrieved from register of the department of sickle cell disease monitoring and management.
Age of study population
No age distinction.
Sex of the study population
Both sexes (female and male) were affected.
Inclusion criteria
Were included in the study, all the people who carried out an electrophoresis of hemoglobin with HOSCO, accompanied or not by a hemogram, whatever their origin.
Exclusion criteria
Subjects with one of the following variables missing from the records: age, sex, outcome.
Hemoglobin electrophoresis
The separation and quantitative estimation of hemoglobin fractions at a basic pH of 9.5 from whole blood was performed using Capillarys 2 Flex Piercing® (Cap 2FP; Sebia, Lisses, France). Briefly, sample diluted in the hemolysis solution is injected into the capillaries. The separation is carried out by the application of a potential difference of several thousand volts across each capillary with direct detection of hemoglobin at 415 nm.
At the end of the analysis, the relative quantification of the fractions is automatically carried out and the profiles can be interpreted visually to detect abnormalities16. The hemogram was performed using automated hematology analyzer (Diatron, Abacus 5, Austria) and Sysmex XN-1000 (Kobe, Japan).
Statistical analyzes
Standard descriptive analysis was performed using EpiInfo 6.04 dfr (CDC, Atlanta, USA) and IBM SPSS 21.0 (SPSS Inc., Illinois, USA) software. One-way ANOVA test was used for comparison of two means while the Tukey multiple comparison test was used more than two means between different groups. The non-parametric Chi-square test (X²) was used to compare the distribution of the different populations and subgroups. The difference was considered significant for the p value ≤ 0.05.
Ethical considerations
This study was approved by the Burkina Faso ethics and health research committee under deliberation number 2018-6-072 of 06 June 2018. The subjects were recruited after free and informed consent and that of legal guardians for kids.
Results
Socio-demographic characteristics
A total of 7,789 people performed hemoglobin electrophoresis and 1014 were associated with hemogram during the period of study. The age of participants ranged from 1 to 40 years old with a mean of 24.86 ± 12.69 years. The subjects consisted of 2361 (30.31 %) male and 5428 (69.69 %) female (Table 1). The age group 16 to 25 ages were the most predominant (38.97 %), while children between 1 to 5 included were the less representative group (7.15 %).
Table 1. Hemoglobin electrophoresis according to sex and age groups
|
Age group (years) |
Male |
Female |
Mean age (years) |
Total |
P value |
|
N (%) |
N (%) |
|
N (%) |
M--ËF |
|
|
≤ 5 |
323 (13.68) |
234 (04.31) |
2.76 ± 1.99 |
557(07.15) |
0.0002 |
|
6 – 15 |
426 (18.04) |
433 (07.98) |
10.92 ± 2.93 |
859(11.03) |
Ë 0.0001 |
|
16 – 25 |
674 (28.55) |
2361 (43.5) |
21.11 ± 2.68 |
3035(38.97) |
Ë 0.0001 |
|
26 – 35 |
612 (25.92) |
1841(33.92) |
29.81 ± 2.71 |
2453 (31.49) |
0.0003 |
|
≥ 36 |
326 (13.81) |
559 (10.3) |
38.46 ± 2.85 |
885 (11.36) |
0.125 |
|
Total |
2361 (30.31) |
5428 (69.69) |
24.86 ± 12.69 |
7789 (100.00) |
Ë 0.0001 |
Biological data
Hemoglobin electrophoresis revealed the following genotypes diagnosed from the 7,789 individuals included in the present study: AA, AC, AS, CC, SC, SS, AE, SO-arab and AO-arab (Fig. 1). Table 2 shows the absolute and relative genotypic frequencies observed and calculated. The AE (3/7789), SO-Arab (2/7789) and AO-Arab (2/7789) genotypes found in seven patients were excluded from statistical analysis. Of the 7,782 hemoglobin electrophoreses, 941 cases of MSCS were observed with a prevalence of 12.09%. Among patients affected with MSCS, the SC genotypes represented 76.30% (718/941) while the SS genotypes were 23.70% (223/941) (Table 2). One quarter of children aged 1 to 5, 25% were carried MSCS including 11.13% SS and 13.82% SC.
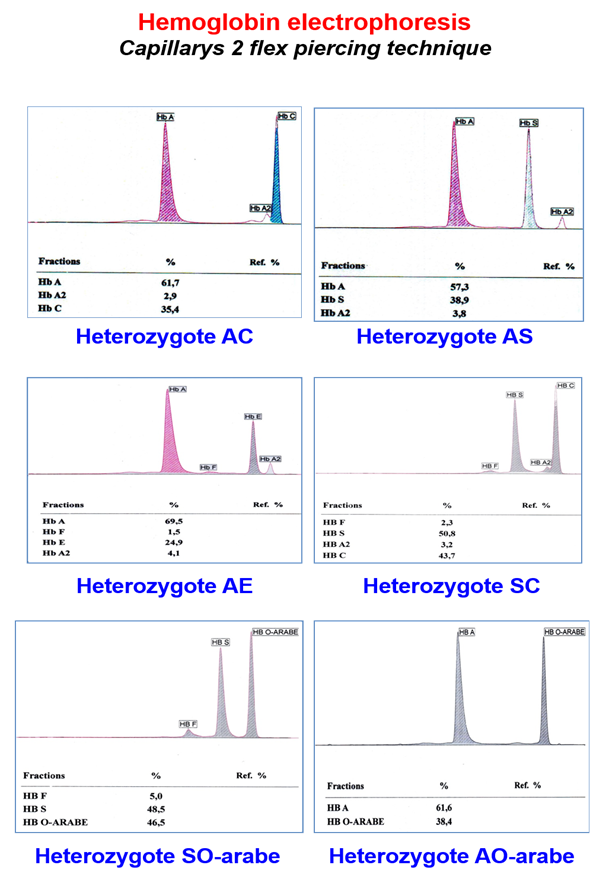
Figure 1: Hemoglobin electrophoretic profiles
Table 2. Observed and calculated genotypic frequencies in the population
|
Genotypes |
Observed absolute genotypic frequencies |
Observed relative genotypic frequencies |
Calculated absolute genotypic frequencies |
Calculated relative genotypic frequencies |
P value |
|
AA |
4575
|
0.5879 |
4076
|
0.5238 |
<0.0001 |
|
AC |
1315
|
0.1690 |
1692
|
0.2174 |
<0.0009 |
|
AS |
799
|
0.1027 |
1420
|
0.1825 |
<0.0001 |
|
CC |
152
|
0.0195 |
175
|
0.0225 |
NS |
|
SC |
718
|
0.0923 |
295
|
0.0379 |
0.003 |
|
SS |
223
|
0.0287 |
124
|
0.0159 |
NS |
|
Total |
7782
|
1.00 |
7782
|
1.00 |
|
|
MSCS |
941
|
0.1209 |
419
|
0.054 |
<0.0002 |
Allelic frequencies were calculated from the observed absolute genotypic frequencies: βA = (2AA+AC+AS)/2N; βC= (2CC+AC+SC)/2N; βS = (2SS+AS+SC)/2N. Absolute genotypic frequencies were determined using the Hardy-Weinberg formula (HW): N (p + q + r)2=Ë AA = Np2; CC = Nq2; SS = Nr2; AC = 2Npq; AS = 2Npr; SC = 2Nqr. p stands for frequency of HbA allele, r for HbS and q for HbC. Table 3 show the allelic frequencies calculated from genotypic data according to age groups; p stands for frequency of HbA allele, r for HbS and q for HbC (Table 3). There was no statistically significant difference in the observed gene frequencies of the HbS and HbC between age groups (Table 3). Noteworthy is the fact that, the frequency of AEs, SO-Arab and AO-Arab genotypes was almost zero due to their small number in the study population. Table 4 presents the hematological parameters such as hemoglobin level, number of red blood cells and mean corpuscular volume (Table 4). On average 64.42% of women aged 1 to 35 were anemic.
Table 3. Allelic frequency of hemoglobin
|
Age group (years) |
Effectif |
βA (p) |
βC (q) |
βS (r) |
(p + q + r)2 =1 |
|
1-5 |
557 |
0.5539 |
0.1858 |
0.2603 |
1 |
|
6-15 |
857 |
0.5694 |
0.1984 |
0.2322 |
1 |
|
16-25 |
3032 |
0.7591 |
0.1382 |
0.1027 |
1 |
|
26-35 |
2452 |
0.7712 |
0.1350 |
0.0938 |
1 |
|
36 à 40 |
884 |
0.7274 |
0.1640 |
0.1086 |
1 |
|
Total |
7782 |
0.7237 |
0.1501 |
0.1262 |
1 |
Table 4. Haematological parameters according to hemoglobin genotypes
|
|
|
RBC (1012/L) |
Hb (g/dL) |
MCV (/fL) |
|
Female (657) |
Mean |
4.21 ± 0.72 |
11.16 ± 1.75 |
79.36 ± 9.66 |
|
Male (357) |
Mean |
4.67 ± 0.90 |
11.93 ± 2.49 |
76.35 ± 9.59 |
|
AA (412)
|
Mean |
4.45 ± 0.65 |
11.98 ± 1.91 |
82.52 ± 8.41 |
|
AC (148)
|
Mean |
4.72 ± 0.70 |
12.00 ± 1.76 |
76.47 ±8.57b |
|
AS (110)
|
Mean |
4.65 ± 0.60b |
12.26 ± 1.73a |
79.95 ± 6.76 |
|
CC (24)
|
Mean |
4.47 ± 0.69 |
10.95 ± 1.68a |
70.65 ± 9.11a |
|
SC (236)
|
Mean |
4.38 ± 0.77a |
10.93 ± 1.68a |
70.81 ± 7.11a |
|
SS (84) |
Mean |
2.97 ± 0.73a |
8.19 ± 1.39a |
81.86± 2.82a |
RBC = Red blood cells; Hb = Hemoglobin; MCV = Mean corpuscular volume
aStudent t-Test p Ë 0.0001
bStudent t-Test p Ë 0.04
Discussion
Sickle cell anemia is an autosomal recessive genetic disorder that affects the beta chain of hemoglobin. It is characterized by an abnormal hemoglobin called hemoglobin S which polymerizes and crystallizes which will result in the stiffening of the red blood cell and its lower deformability. There are several forms of sickle cell disease (SS, SC, S Beta, etc.). It is a chronic hemolytic disease manifested by: severe anemia, severe bacterial infections and iso-vaso-occlusive ischemic attacks (AVO). The diagnosis is based on the electrophoresis of hemoglobin.
This retrospective study carried out at Saint Camille Hospital in Ouagadougou (HOSCO) revealed that the population of the current study varied from 1 to 40 years with an average of 24.86 ± 12.69 years.
Among them, 7.15% were no more than 5 years of age and women participant for hemoglobin electrophoresis outnumber men (69.69% vs. 30.31%). This high percentage of women participant could be linked to prenatal follow-up at HOSCO’s Mother and Child Health service. It is noteworthy that the predominant age group 16 to 35 years (70.46%) corresponds to the age of procreation. These results are consistent with those of Guédéhoussou et al.13 in Togo, whose ¾ of the respondents were between 20 and 39 years old and represent the most fertile age group with a female predominance and those of Douamba et al.11 with more women (51.9%) than men surveyed (48.1%). Guédéhoussou et al.13 linked this large number of women to the absence of men at the time of the interview for professional reasons. On the other hand, Diarra et al.17, in Burkina Faso as well as Diop et al.18, in Senegal reported more men than women in their studies. In their work, the sex ratios were respectively 1.44 and 1.25. This difference in sex ratio from one study to another would be linked to the choice of the study population.
The present study reported 718 SC genotypes versus 223 SS genotypes. These results are in line with the literature describing more SC carrier than SS individual including Simpore et al.12 with 660 SC and 196 SS recorded, Ayéroué et al.19, 38 SC and 23 SS as well as Diarra et al.17, 47 SC and 29 SS. It is noteworthy that the relative genotypic frequency of SC is higher than that of SS in west Africa (0.092 versus 0.029) (Table 2) probably because clinical and hematological parameters of heterozygous HbSC individuals are less critical compared to homozygous HbSS14.
However, some studies report more HbSS subjects than HbSC individuals19,20. These differences could be due to the Burkinabe origin of HbC15,21 and the demographic data of each country because the sickle cell disease is not a X-linked inherited disease8. HbC is the most common abnormal hemoglobin in West Africa with its epicenter located in Burkina Faso (in the Mossi plateau, Bissa country), northern Ghana, northern Benin and northern Togo21. From the results obtained, it is noted that the allelic frequency of βC is higher than that of βS (0.150 of βC against 0.126 for βS). These results are consistent with those of Ayéroué et al.19 (2009) and Ouedraogo et al.10. No statistically significant difference was found in the βC and βS allele frequency between the different age groups. The observation in West Africa is that there is more common SC type of MSCS compared to the SS genotypes19,22. This predominance of SC could be explained by the early mortality of SS individuals (vulnerable age group of 1 to 5 years)19. Our results show that 25% of children from 1 to 5 years old were diagnose with MSCS including 11.13% SS and 13.82% SC.
Simpore et al.14 reported a significant difference between MSCS and the other genotypes in children. We also compared the results of this study with those of Simpore et al., in 200214 who had also carried out their work in HOSCO. The distributions between the genotypes have a difference which is statistically significant (p Ë 0.0001). These results, which show that the two populations are not in equilibrium according to the Hardy - Weinberg Equilibrium Statistics Test distributions, more probably testify to the current long life of sickle cell patients treated with HOSCO but also to the effectiveness of sensitization of the public who would justify the increased number of holy carriers (AS and AC) diagnosed (Table 5). Note that over the years, in Burkina Faso, for MSCS, there has been an improvement in the supply of care at all levels especially the effective implementation of neonatal screening, the training of health workers, the development of treatment protocols, public awareness.
Table 5. Prevalence of hemoglobin genotypes compared with the results of Simpore et al.21
|
Genotypes |
TRAORE et al., HOSCO 2019 |
SIMPORE et al., HOSCO 2003 |
|
|
N (%) |
N (%) |
|
AA |
4575 (58.74) |
5910 (58.13) |
|
AC |
1315 (16.88) |
1960 (19.28) |
|
AS |
799 (10.26) |
1249 (12.29) |
|
CC |
152 (1.95) |
191 (1.88) |
|
SC |
718 (9.22) |
660 (6.49) |
|
SS |
223 (2.86) |
196 (1.93) |
|
AE |
003 (0.04) |
000 (0.00) |
|
AO-arab |
002 (0.03) |
000 (0.00) |
|
SO-arab |
002 (0.03) |
000 (0.00) |
|
Total |
7789 (100.00) |
10166 (100.00) |
X2: Traore --Ë Simpore : p Ë 0.0001
Here we report three new genotypes compared in Burkina Faso namely AE genotype in three (03) people aged 13, 25 and 28 respectively, the AO-Arab genotype in two (02) people aged 20 and 40, and the SO-Arab genotype in two (02) people 6 and 23 years old. According to the literature, HbE geographic origin is Asia while HbE-Arab originated from Africa9. These new AE, AO-Arab and SO-Arab genotypes diagnosed could explained by the increase migratory flow and the geographical spread of certain genes such as HbE and HbO-Arab4,15, but also the advanced techniques of diagnostic methods. Unlike Simpore et al (2002) who used cellulose acetate plate electrophoresis, a technique that lacks precision, the present study used a new technique, capillary electrophoresis. With these new techniques like high performance liquid chromatography (HPLC) and capillary electrophoresis, we manage to separate the hemoglobin and allow a good quantification of hemoglobin A2 and hemoglobin F. Thus, the HbO-arab detaches in a fine band between HbA and HbS, while HbC has a more cathodic migration than HbS and HbE co-migrates with HbA9,23(Fig 1).
By comparing the SS and SC individuals, the SS individuals had the lowest number of red blood cells (GR), 2.97 ± 0.73 million/mm3 compared to 4.38 ± 0.77 million/mm3 for SC. The same is true for the hemoglobin (Hb) level 8.19 ± 1.39 g/dL in the SS against 10.93 ± 1.68 g/dL for the SC. The hemogram shows us anemia in subjects with MSCS. This is in line with the results of Simpore et al.12. According to the literature, SS individuals had moderate anemia with an Hb level = 8.19 g/dL6; while SC had less pronounced moderate anemia (Hb level = 10.93 g/dL)2. For the Mean corpuscular volume (MCV), it was 81.86 ± 12.82 fL in the SS versus 70.81 ± 7.11 fL for the SC. 0.51 % microcytic anemia was observed among the people who performed the hemogram (Table 4). SS have moderate, microcytic anemia if the margin of error is considered. The work of Thiam et al.8 also noted that all SS children were anemic but only 50 % had microcytic anemia and the other 50 % had normocytic anemia (Thiam et al., 2017).
In the study of Dahmani et al.6 88.5 % of the SS diagnosed were anemic. Their results showed a very variable degree of anemia with 67.8 % normocytic anemia, against 29.9 % microcytic anemia and 2.3 % macrocytic anemia. Indeed, Gbadoé et al.24 diagnosed macrocytic anemia in the SS and Olivier et al.9 normal and even macrocytic anemia. People with CC hemoglobins do not have sickle cell anemia. They don't have a major health problem. The results of the hemogram show it with 4.47 ± 0.69 million / mm3 of red blood cells (GR), the hemoglobin (Hb) level 1.95 ± 1.68 g / dL and the average red blood cell volume (VGM) 70.65 ± 9.11 fL. On the other hand, there is a microcytic anemia as in all the groups carrying the βC allele.
At HOSCO, children aged 1 to 5 who have performed the hemogram are not all carriers of MSCS (only 0.53 % of the study population). However, anemia is observed at 96.35% in this age group (Table 4). It is an age group vulnerable to infections such as malaria responsible for severe anemia in children and deaths in sickle cell anemia at this age were due to severe anemia (47.8%)25. Among women aged 1 to over 35, an average of 64.42% are anemic. The highest cohort of women with anemia is between 16 and 25 years of age (68.27%) and between 26 and 35 years of age (66.49%). The statistical difference between these 2 age groups and the others is indeed significant. Women at this age are likely to procreate. However, the causes of worsening anemia in women may be linked to pregnancy26.
Conclusion
Sickle cell anemia is a hemoglobinopathy which is a serious public health disease. From this study, it appears that the prevalence of HbC and HbS hemoglobinopathies is high in Burkina Faso. Indeed, the absolute genotypic frequencies and the allelic frequencies of patients with MDS are on the increase, given the awareness and accessibility of medical care for sickle cell patients. The study made it possible to diagnose for the first time with the improvement of the technical platform of the laboratories, the AE, AO-Arabic, SO-Arabic genotypes at HOSCO, Burkina Faso.
Failing to eradicate it, sickle cell disease must be followed to reduce its impact on the population. And to do this, it is necessary to go through public awareness, prevention, early detection of the disease and medical monitoring of sickle cell anemia.
Does the HbC allele protect against malaria infection? it would be interesting to conduct studies along these lines.
List of abbreviations
|
ANOVA |
Analysis of variance |
|
MCV |
Mean corpuscular volume |
|
CERBA |
Pietro Annigoni Biomolecular Research Center |
|
fL |
Femtolitre |
|
Hb |
Hemoglobin |
|
HbC |
Hemoglobinopathies C |
|
HbE |
hemoglobinopathies E |
|
HbS |
Hemoglobinopathies S |
|
HOSCO |
Saint Camille Hospital of Ouagadougou |
|
HPLC |
High-performance liquid chromatography |
|
LABIOGENE |
Laboratory of Molecular Biology and Genetics |
|
MSCS |
Major sickle cell syndrome |
|
pH |
Potential of hydrogen |
|
SPSS |
Statistical Package for the Social Sciences |
|
USTA |
University Saint Thomas d'Aquin |
|
RBC |
Red blood cells
|
Authors' contributions
MST and JS conceived and designed the study. MST, TMZ, AKO, GG, MB and PO were involved in data generation, collection and assembly. GLM, IT and AKO were involved in data analysis and interpretation. MST, AKO, DO-Y, JS, TB, FS and JS were involved with drafting or revising the manuscript. MST, TMZ, IT, PO and JS provided administrative, technical and material support. Supervision of the study was made by MST, TMZ, PO and JS. All authors critically revised and approved the final version of this publication.
Acknowledgements
The authors would like to thank the Hospital of Saint Camille (HOSCO) and CERBA/LABIOGENE for their collaboration in carrying out this research on hemoglobinopathies in Burkina Faso.
References
- Jahangiri M. Decision-tree-based methods for differential diagnosis of β-thalassemia trait from iron deficiency anemia 2017. Expert Systems. 34(3): 1–6. https://doi.org/10.1111/exsy.12201
- Mattioni S. La drepanocytose en France. Revue Francophone des Laboratoires. 2016; 2016(481): 61-66.
- Vasseur C, Baudin-Creuza V. Role du chaperon moléculaire de l’alpha-hémoglobine dans la formation de l’hémoglobine et l’expression clinique de certaines hémoglobinopathies. Transfusion Clinique et Biologique. 2015; 22(1): 49-57.
- Labie D, Elion J. Modulation polygénique des maladies monogéniques: l'exemple de la drépanocytose. Med Sci (Paris). 1996; 12(3): 341-9.
- Krishnan K, Martinez F, Wille RT, et al. HB Washtenaw [βT11(A8)VAL-PHE]: An Electrophoretically Silent, Unstable, Low Oxygen Affinity Variant Associated with Anemia and Chronic Cyanosis. Hemoglobin. 1994; 18(4-5): 285-295.
- Dahmani F, Benkirane S, Kouzih J, et al. Evaluation of hemogram in patients with homozygous sickle cell disease: about 87 cases. The Pan African medical journal. 2016; 25: 240-240.
- WHO. Drepanocytose une strategie pour la Region africaine de l’OMS rapport du Directeur regional. [Technical documents] 2011 2011-05-27 02-03-2020]; Available from: https://apps.who.int/iris/handle/10665/1727.
- Thiam L, Drame A, Coly IZ, et al. [Epidemiological, clinical and hematological profiles of homozygous sickle cell disease during the intercritical period among children in Ziguinchor, Senegal]. Pan Afr Med J. 2017; 28: 208.
- Oliver M, Wolf A, Roche C, et al. [Hemoglobinopathy. Laboratory diagnosis]. Med Trop (Mars). 2011; 71(3): 217-22.
- Ouedraogo S. Evaluation du cout medical direct de il prise en charge du syndrome drepanocytaire majeur de l'enfant a Ouagadougou. Science et Technique, Sciences de la Sante. 2013; 36(1-2): 73-82.
- Douamba S, Nagalo K, Tamini L, et al. Major sickle cell syndromes and infections associated with this condition in children in Burkina Faso. The Pan African medical journal. 2017; 26: 7-7.
- Simpore J, Pignatelli S, Barlati S, et al. Biological and clinical presentation of patients with hemoglobinopathies attending an urban hospital in Ouagadougou: confirmation of the modification of the balance between Hb S and Hb C in Burkina Faso. Hemoglobin. 2002 a; 26(2): 121-7.
- Guedehoussou T, Gbadoe AD, Lawson-Evi K, et al. Connaissance de la drépanocytose et pratiques de prevention dans la population d’un district urbain de Lome, Togo. Bul Soc Pathol Exot. 2009; 102: 247-251.
- Simpore J, Pignatelli S, Barlati S, et al. Modification in the frequency of Hb C and Hb S in Burkina Faso: an influence of migratory fluxes and improvement of patient health care. Hemoglobin. 2002 b; 26(2): 113-20.
- Simpore J. Anthropological consideration on prevalence and fitness of β C and β S genotypes in Burkina Faso (a survey in the public schools). International Journal of Anthropology. 2002 c; 17(3): 139-151.
- Jeanne L. Place de l’électrophorese capillaire dans le diagnostic et le suivi des hémoglobinopathies. Option/Bio. 2010; 21(434): 17-20.
- Diarra Y, Koueta F, Dao L, et al. Prise en charge de la drépanocytose en milieu pédiatrique : expérience du centre hospitalier universitaire pédiatrique Charles-de-Gaulle de Ouagadougou (Burkina Faso). Cahiers d'études et de recherches francophones / Santé. 2008; 18(2): 71-75.
- Diop S, Mokono SO, Ndiaye M, et al. La drépanocytose homozygote après l’age de 20 ans : suivi d’une cohorte de 108 patients au CHU de Dakar. La Revue de Médecine Interne. 2003; 24(11): 711-715.
- Ayeroue J, Kafando E, Kam L, et al. Le syndrome drépanocytaire de type hémoglobine SC : expérience du CHU Yalgado Ouedraogo de Ouagadougou (Burkina Faso). Archives de Pediatrie. 2009; 16(4): 316-321.
- Diagne I, Ndiaye O, Moreira C, et al. Les syndromes drepanocytaires majeurs en pédiatrie a Dakar (Senegal). Archives de Pediatrie. 2000; 7(1): 16-24.
- Labie D, Richin C, Pagnier J, et al. Hemoglobins S and C in Upper Volta. Hum Genet. 1984; 65(3): p. 300-2.
- Simpore J. Prevalence des hémoglobinopathies HbS et HbC au Burkina. Burkina medical. 2003; 32: 1-13.
- Mario N, Sala N. Diagnostic biologique des hemoglobinopathies. Revue Francophone des Laboratoires. 2016; 2016(481): 35-47.
- Gbadoe A, Atsou K, Agbodjan-Djossou OA, et al. Prise en charge ambulatoire des drépanocytaires: evaluation de la premiere année de suivi des patients dans le service de pediatrie de Lome (Togo). Bull Soc Pathol Exot. 2001; 94(2): 1001-105.
- Koko J, Dufillot D, M'Ba-Meyo J, et al. Mortalite des enfants drepanocytaires dans un service de pediatrie en Afrique Centrale. Archives de Pediatrie. 1998; 5(9): 965-969.
- Elira Dokekias A, Ngolet Ossini L, Atipo Tsiba FO, et al. Evaluation de la transfusion sanguine chez 112 patients drepanocytaires homozygotes au CHU de Brazzaville. Transfusion Clinique et Biologique. 2009; 16(5): 464-470.