
Coats Plus Syndrome and Mutation of the TERT Gene: A Case Report
Typhaine Grenet1,2, Caroline Kannengiesser3,4,5, Raphael Borie3, 5, 6, 7, Audrey Giocanti-Aurégan2
1Centre d’imagerie et de Laser, 11 rue Antoine Bourdelle, Paris, France
2Ophthalmology department, Avicenne hospital, Paris 13 University, APHP, Bobigny, France
3Université Paris Diderot, Paris, France
4Laboratoire de Génétique, APHP, Hôpital Bichat, Paris, France
5Service de Pneumologie A, Centre de Compétence des Maladies Pulmonaires Rares, Paris, France
6DHU FIRE, Hôpital Bichat, APHP, Paris, France
7INSERM, Unité 1152, Paris, France
Abstract
Purpose: To report a case of bilateral Coats Plus syndrome associated with a TERT gene deletion.
Case report: A 35-year-old man with a history of liver transplant, hematological impairment, and lung fibrosis due to a TERT gene mutation was referred for blurred vision in the right eye (RE). Ophthalmological examination revealed bilateral peripheral ischemia of the retina associated with bilateral peripheral telangiectasia and telangiectasia at the posterior pole of the RE responsible for macular edema, confirmed by multimodal imaging.
Conclusion: In this young patient with bilateral Coats-like lesions associated with systemic liver and lung failures due to a TERT mutation, we report a case of Coats plus syndrome due to a TERT mutation.
Case Report
A 35-year-old man was referred for blurred vision in his right eye (RE).
Visual acuity was 20/40 in the RE and 20/20 in the left eye (LE). The slit lamp examination was unremarkable. Intraocular pressure was normal in both eyes. The fundus examination revealed hard exudates and hemorrhages at the right posterior pole (Fig.1A) and bilateral temporal peripheral vascularization rarefaction (Fig. 1 A, B). Optical coherence tomography (OCT) of the RE revealed a voluminous macular edema (Fig. 2A, B), and OCT of the LE was normal (Fig. 2C). Fluorescein angiography (Fig. 3A-D) showed bilateral retinal ischemia of the temporal periphery with bilateral capillaropathy and telangiectasia in the temporal periphery. In the RE (Fig. 3A, B), telangiectasia were also observed at the posterior pole, responsible for fluorescein leakage in the late frames (Fig. 3B). A bilateral hyperfluorescence of the optic disc was also seen (Fig. 3B, D). OCT angiography confirmed the presence of telangiectasia dilation in the right fovea, responsible for macular edema (Fig. 4 A-D) and peripheral vascularization rarefaction (Fig. 4 E).
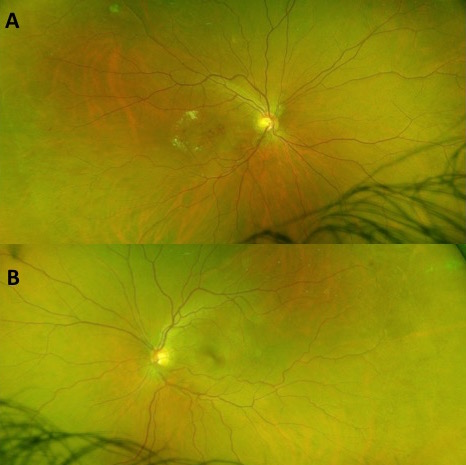
Figure 1: Color retinal photographies of the right (A) and left (B) eyes showing bilateral temporal peripheral vascularization rarefaction. Exudates and hemorrhages of the right posterior pole (A).
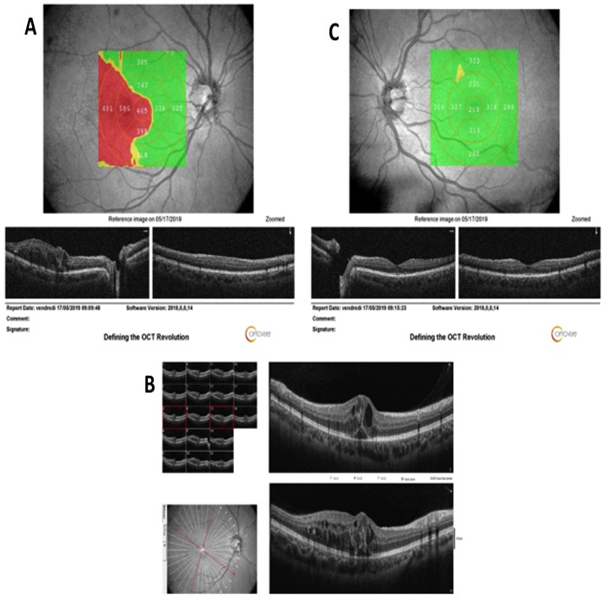
Figure 2: Horizontal OCT B-scans of the right (A) and left (B) eyes. A macular edema with a central retinal thickness of 465 microns is seen in the RE, while the LE is normal. High definition OCT of the RE (C) shows the cystoid macular edema associated with serous retinal detachment.
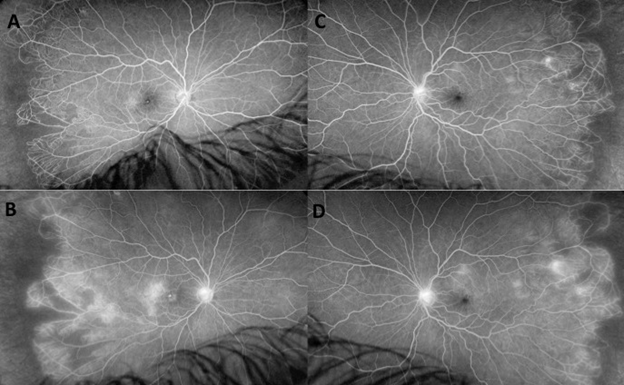
Figure 3: Fluorescein angiograms of the right (A, B) and left (C, D) eyes showing bilateral retinal ischemia of the temporal periphery with bilateral capillaropathy and telangiectasia in the temporal periphery. In the RE, telangiectasia are also seen at the posterior pole, responsible for fluorescein leakage in the late frames (B). A leakage from peripheral telangiectasia and optic disc hyperfluorescence are also observed in the late frames (B, D) compared to the early (A) and intermediate frames (C).
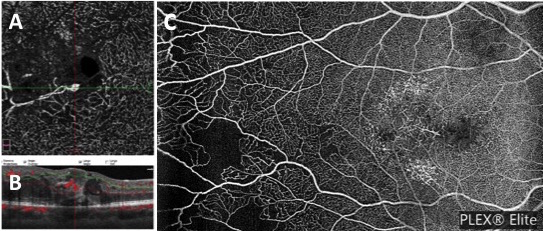
Figure 4: OCT angiography of the RE showing telangiectasia dilation in the right fovea (A, B) responsible for macular edema. OCTA of the temporal periphery confirmed the vascularization rarefaction (C).
The patient had long personal medical history since he had early hair greying since the age of 13, he underwent a liver transplant 8 months ago for liver fibrosis, and he also had pancytopenia and idiopathic lung fibrosis (IPF). His father died 3 years ago of IPF. A diagnosis of telomeropathy responsible for these systemic failures was suspected in this family with familial IPF, liver disease and bone marrow failure, A molecular exploration was performed on the DNA of the father. A germline heterozygous missense variation was identified in exon 6 of TERT: c.2225G>A, p.Arg742His Our patient was also carrier of this variation confirming a telomeropathy transmitted in an autosomic dominant manner. The missense variation of TERT was interpreted as pathogenous (class 5) due to its rare frequency, the in silico analysis predicting a deleterious effect, co segregation of the variation with the phenotype and the identification of this variation in another independent family (our unpublished data, C Kannengiesser and the publication recently in another patient with IPF1.)
Based on bilateral Coats-like lesions associated with systemic involvement, we suggested the diagnosis of Coats Plus syndrome (CPS) due to the TERT gene pathogenous variation.
Discussion
CPS also referred to as cerebro-retinal microangiopathy with calcifications and cysts (CRMCC)2,3 is a genetic disease that was described in 1988 by Tolmie et al4. The prevalence of CPS is less than 1 case for 1 billion. It has been first described in association with CTC1 (conserved telomere maintenance component 1) bi-allelic mutations. This gene encodes a protein participating to the maintenance of the integrity of the telomeres, the TTAGGG repeated sequences at the ends of the chromosomes. The transmission is autosomal recessive. The ophthalmologic lesions described were similar to Coats disease, an idiopathic disorder characterized by an abnormal development of retinal vessels (telangiectasia) with progressive exudation leading potentially to exudative retinal detachment, although they were bilateral. The ophthalmic involvement is bilateral in CPS while only 25%5,6 to 62%7 of cases of classical Coats disease show a bilateral involvement. Another difference is that in CPS, a systemic involvement is associated, unlike in Coats disease. The two main ophthalmologic complications of Coats disease and CPS are retinal neovascularization and exudation.
CPS has also been associated to another telomeropathy due to a TERC deletion8. Mammalian telomeres are maintained by an enzyme referred to as telomerase, a complex that includes a RNA template (hTERC), a reverse transcriptase subunit (hTERT) ‘coded by’ TERC and TERT, and various other proteins. This enzyme prevents a progressive telomere shortening8. Critical telomere shortening results in a proliferative defect in hematopoietic stem cells leading to bone marrow failure syndrome9.
The exact pathophysiological mechanism underlying Coats disease is unknown, while we know that CPS is part of the spectrum of telomeropathies. Several patients with CPS have been shown to have shortened telomeres, suggesting that telomere dysfunction could play a role in CPS pathogeny10.
The case described by Peene et al.8 supported this assumption and they have suggested that the presence of bone marrow failure and telomere dysfunction in patients with CPS could strongly support a relationship between the disruption of telomere homeostasis and the ocular pathology and that the TERC gene deletion could be etiologically associated with exudative retinopathy.
Our case is the first description of CPS associated with a TERT gene mutation. This could be due to the fact that our patient had a particularly severe form of telomeropathy with lung fibrosis11 and that he had visual symptoms only due to the presence of the right macular edema. It could be assumed that in the presence of extra central exudation, the patient would have remained visually asymptomatic.
Regarding ophthalmological treatment, exudations are usually treated with photocoagulation of the telangiectasia vessels responsible for the exudation5,6. In our case, photocoagulation was not an option given the central location of the telangiectasia dilation responsible for edema. It has thus been decided to treat the RE with anti-VEGF injections.
In conclusion, in a young patient with bilateral Coats-like lesions associated with systemic liver and lung failures due to a TERT mutation, we described a case of CPS. Ophthalmological examination should be carried out in telomeropathy individuals.
References
- Petrovski S, Todd JL, Durheim MT, et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. Am J Respir Crit Care Med. 2017; 196: 82–93.
- Polvi A, Linnankivi T, Kivelä T, et al. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am J Hum Genet. 2012; 90: 540–549.
- Crow YJ, McMenamin J, Haenggeli CA, et al. Coats’ plus: a progressive familial syndrome of bilateral Coats’ disease, characteristic cerebral calcification, leukoencephalopathy, slow pre- and post-natal linear growth and defects of bone marrow and integument. Neuropediatrics. 2004; 35: 10–19.
- Tolmie JL, Wilcox DE, McWilliam R, et al. Palmoplantar keratoderma, nail dystrophy, and hereditary motor and sensory neuropathy: an autosomal dominant trait. J Med Genet. 1988; 25: 754–757.
- Sigler EJ, Randolph JC, Calzada JI, et al. Current management of Coats disease. Surv Ophthalmol. 2014; 59: 30–46.
- Troumani Y, Ackermann F, Cohen S, et al. [Coats Plus: Coats disease with systemic features]. J Fr Ophtalmol. 2016; 39: e167-170.
- Blair MP, Ulrich JN, Elizabeth Hartnett M, et al. Peripheral retinal nonperfusion in fellow eyes in coats disease. Retina Phila Pa. 2013; 33: 1694–1699.
- Peene G, Smets E, Legius E, et al. Unilateral Coats’-like disease and an intragenic deletion in the TERC gene: A case report. Ophthalmic Genet. 2018; 39: 247–250.
- Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012; 13: 693–704.
- Shields JA, Shields CL. Review: coats disease: the 2001 LuEsther T. Mertz lecture Retina Phila Pa. 2002; 22: 80–91.
- Borie R, Kannengiesser C, Sicre de Fontbrune F, et al. Management of suspected monogenic lung fibrosis in a specialised centre. Eur Respir Rev Off J Eur Respir Soc. 2017; 26.