
Yeast contributions to Alzheimer’s Disease
Jamieson B Mcdonald, Sudip Dhakal, Ian G Macreadie*
School of Science, RMIT University, Bundoora, Victoria, Australia
Abstract
Alzheimer’s Disease is a highly prevalent, age-related, neurodegenerative disease associated with the accumulation of toxic proteins, including amyloid beta and tau, that affect important cellular functions. Through the study of these proteins in yeast over the past 2 decades, the effects of amyloid beta oligomerization and aggregation, and tau hyperphosphorylation on basic cellular functions, such as ageing, oxidative stress, cell cycling and proteostasis have been observed. Strategies for the prevention of damage by these proteins can be explored, thanks to the exquisite array of technologies available for yeast studies. This review summarises existing knowledge of Alzheimer’s Disease, the work over the past two decades on yeast models for Alzheimer’s Disease and how these models contribute to the development of treatments and preventative strategies for Alzheimer’s Disease.
Introduction
Alzheimer’s disease
Globally, 50 million cases of cognitive impairment are associated with Alzheimer’s disease (AD), the most common form (60-80%) of dementia and major cause of death in elderly people. This number is expected to triple to 152 million by 20501,2,3. In addition to AD’s profound impact on quality of life, the disease is also a major healthcare, economic and social burden. It was reported in America alone, in 2018, an estimated 18.5 billion hours of care was provided by unpaid care workers and family members for those suffering from AD2. This care increases the risk of emotional distress and adverse mental and physical health consequences for family members and healthcare workers, which is valued at approximately $234 billion in the US alone2. Long-term care, health care, and hospital services for people over the age of 65 with dementia accounted for an estimated $290 billion in 20192. The associated healthcare costs of the disease are estimated to exceed $1 trillion by 20504.
AD mostly affects people above the age of 65 (late-onset AD), however, people as young as 40-50 years can develop genetically inherited, early-onset familial AD1. AD is a progressive neurodegenerative brain disorder that starts to develop years before symptoms arise2. During AD progression, disrupted cellular functions, loss of cellular homeostasis, and impaired cellular defenses cause significant irreversible structural and functional impairment to healthy brains leading to the death of neurons1,3. Damage to the hippocampus and neocortex leads to memory loss, language difficulties and learning defects4. Further, damage as AD progresses may result in a decline in other cognitive areas resulting in the complete incapacity to perform essential daily activities and the ability to function independently4.
AD is characterized by the extracellular accumulation of toxic amyloid beta (Aβ) plaques and intracellular hyperphosphorylation of tau in neuronal cells, oxidative stress, mitochondrial dysfunction, and neuronal atrophy1,3. Research has led to the development of several hypotheses to address the molecular mechanisms of AD5. However, the connections between these factors and neurodegenerative disorders remains elusive and the roles of the complex components of the disease has been disputed for decades. Despite, improved understanding of AD pathogenesis through the utilisation of yeast models and mammalian studies, gaps remain regarding the exact molecular changes and biological processes in the brain that lead to AD. These knowledge gaps present significant challenges in developing effective AD treatments. Presently there are no therapeutics prescribed for the prevention or alleviation of AD progression despite ~200 clinical trials in the past two decades searching for treatments.
The amyloid beta hypothesis represents the leading explanation for AD pathogenesis thus there is widespread research to inhibit Aβ-induced damage and subsequent death of neuronal cells6. Aspects of AD can be studied using numerous methods including ex vivo and in vitro experimental tools, computational studies, and transgenic models5. Approaches to reduce Aβ-induced AD focus on the inactivation or removal of Aβ via antibodies, and strategies to block the formation of toxic oligomers and plaques7. Translational outcomes have been constrained, owing to the shortcomings of animal models and their assay methods, including the high throughput screening of potential drugs and chemo preventatives7. There is, therefore, a need to adopt models that enable high throughput screening methods to accelerate the discovery and development of new treatment strategies. Yeast as a eukaryotic model organism has been utilized to address such limitations and has clearly demonstrated its effectiveness in studying the molecular aspects of AD.
This review article focuses on yeast’s pioneering contributions to AD research, and the recent insights and developments yeast has played in the study of AD. Emphasis is placed on the significant contributions yeast has made to translational research in the pre-screening of compounds that could be used to treat and/or prevent AD. Additionally, the review will highlight the way forward in using yeast model systems to provide further insights on the mechanisms of AD and for identifying multifactorial drugs or therapeutics that target AD.
Yeast as a model organism to study Alzheimer’s disease
The budding yeast, Saccharomyces cerevisiae, is the most popular unicellular model organism and has been used as a model for most AD studies described here. It was the first eukaryotic organism to have its genome fully sequenced and the information derived has facilitated substantial knowledge on cell biology relevant to humans. S. cerevisiae contains 6000 genes and ~31% have a homolog in humans4,8. The short generation time of yeast, ease of culture and its ability to be readily transformed to produce human proteins in a desired context make it an efficacious model for studying chronic diseases such as AD. Although yeast lack specific processes of neuronal cells, a nervous system, immune system, and neuropathology associated with cell-cell communications, the majority of the molecular signaling pathways and proteins involved in human neurological diseases exhibit conserved sequences and function9,10. A large fraction of essential cellular processes, including cell cycle progression, mitochondrial biology, metabolism, transcription, translation, protein secretion, protein quality control mechanisms, vesicle trafficking and apoptosis are highly conserved between this unicellular fungus and humans, including our neuronal cells4, providing an efficacious platform to study protein-misfolding pathologies11. Hence, S. cerevisiae provides a facile means of studying both physical and genetic associations between proteins and simple cellular mechanisms, paving the way for the utilisation of yeast in medicinal and medical research. Yeast have played an essential role in the development of many high-throughput techniques including DNA and ChIP-chip microarrays, in the large scale analysis of protein-protein interactions (yeast two-hybrid and three-hybrid systems), transcriptomics, proteomics and metabolomics analyses9.
Ageing is an inevitable factor of life and an underlying cause of numerous chronic diseases such as AD. Ageing is a multifactorial process involving the build-up of molecular, cellular, and systemic damage leading to physiological impairment and a decline in reproductive ability and survival12. Ageing humans exhibit very similar features to ageing S. cerevisiae (that are identified by 2 or more bud scars) such as accumulation of aberrant proteins, reduced telomeres, oxidative damage and mitochondrial dysfunction10. An advantage of using yeast for AD research is that it is less complex compared to mammalian models and affords a relatively simple means to investigate elements associated with AD. In addition, the use of S. cerevisiae enables large scale genetic screening and functional genomic studies that are not achievable in humans. Furthermore, because the budding yeast displays similar cellular aspects of ageing humans it can be utilised as a model to screen compounds that reinstate health to old cells and to identify potential therapeutics. As a result, yeast models have provided unprecedented insights into the underlying molecular basis of ageing and to decipher the complexity of disease pathology and disease associated mutations such as those involved in AD.
All the features discussed above make yeast an efficacious system for genome-wide screening of genes that affect lifespan or the toxicity of heterologously expressed human proteins such as Aβ and tau. In addition, the investigation of gene and protein interaction networks regulating longevity and ageing are achievable. Yeast have been extensively utilized as a model organism for studying AD (Figure 1) and neurodegeneration as discussed in some previous reviews4,5,7,10.
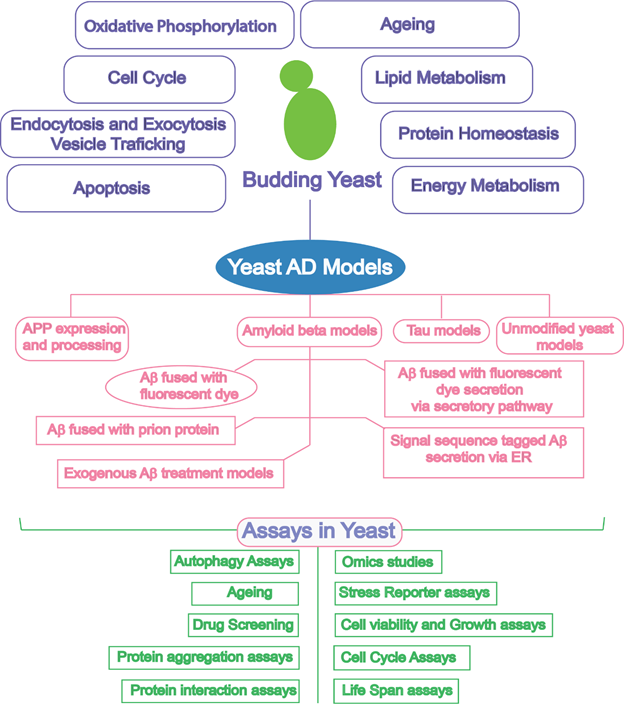
Figure 1: Schematic diagram outlining the biological similarity of yeast and research tools offered by yeast to enable exploration of yeast as a model for Alzheimer’s disease. AD, Alzheimer’s disease; APP, amyloid precursor protein; Aβ, amyloid beta; ER, endoplasmic reticulum.
Although Aβ and tau have no orthologs in yeast, their heterologous expression is readily achieved and can be highly informative in understanding the pathobiology of these proteins4. Yeast models have contributed to understanding the pathology of the disease via modelling amyloid precursor protein (APP) processing, in vivo Aβ oligomerization, Aβ-associated toxicity, and tau phosphorylation10. Table 1 shows the contributions of yeast cell-based assays to the study of AD and recent advances in the use of yeast.
Table 1. Yeast AD Models with main findings and insights
|
Amyloid Precursor Protein (APP) expression and processing models
|